- 1) When I upload my own data, why do I get an error message
saying the input format is wrong?
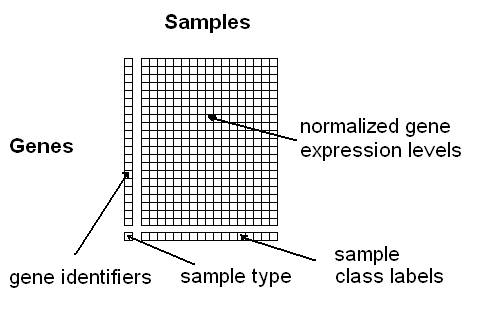
If you haven't downloaded
the example input file in the "Data Set" section, please try this
first. A typical problem is that users forget to specify the
class labels in the last row of the input; however, this is only
required on the supervised analysis modules and not on the
Class Discovery module. Moreover, please note that ArrayMining.net
currently does not provide missing value imputation for your data (this
is mainly because we don't know whether these values are missing
completely at random in your data or depend on other variables).
To specify the class labels either integers or strings can be
used, only continuous values are not supported (not that on the Class
Discovery module the last input row will be interpreted as
an additional data row, if it contains continuous values,
and no adjusted rand indices will be computed).
- 2) Why do I only obtain annotations for a subset of the genes
on the Gene Selection Analysis module?
For many probes on a
microarray a corresponding gene is either not (yet) annotated in
public data bases or does not exist (probes do not necessarily only
represent genes). Moreover, different array platforms use different
gene identifiers and ArrayMining.net does not yet support automatic
gene name normalization. If you use standard identifiers (e.g.
ENTREZ ID, GENBANK ACCESSION, etc.) most of the gene names should
be recognized - otherwise, you can use an external public gene name
conversion service on the web. We recommend the DAVID service (http://david.abcc.ncifcrf.gov
), the CNIO Clone/Gene ID converter (http://idconverter.bioin
fo.cnio.es) and the MIPS CRONOS service (http://mips.
gsf.de/genre/proj/cronos/batch.html).
- 3) Which platforms are supported by ArrayMining.net?
Our
system supports pre-normalized from any human microarray-platform -
the only condition is that your data is submitted as matrix with
columns corresponding to samples and rows corresponding to genes.
However, if you want to use the functional annotation features, your
data must contain one gene identifiers in one of the supported
formats (see Instructions and question 2).
- 4) On the Gene Set Analysis module, why do I always obtain
the message that none of the gene sets are enriched within my data
or that the gene labels don't match?
If you don't get any
results on the Gene Set Analysis module this can have multiple
reasons: Your gene labels might not be in one of the standard
formats (ENTREZ ID, GENBANK ACCESSION, etc.), the genes might not be
contained in the gene sets (this is unlikely in the case of the KEGG
and GO gene sets, but very likely in the case of the cancer-related
gene sets) or no gene set has passed the test for statistical
significance of enrichment in your data (default: q-value <
0.05).
In order to rule out that the wrong gene identifiers have
been used, you can submit your gene names to an external public gene
name conversion tool (see question 2 for examples).
- 5) Why can I use the GEO data sets only on the Class
Discovery module?
Supervised analysis is currently not supported
for the GEO data base, although some GEO entries contain labelled
data. The reason for this is simply that the label information is
not standardized and can not be extracted automatically. Thus, for a
supervised analysis of GEO data sets, the users needs to download
the data first on his own computer and specify the class information
manually before uploading it on ArrayMining.net.
- 6) On the Class Assignment Analysis module, why are the
standard deviations so high?
This is a usual and hard to solve
problem for small-sample microarray data, especially if leave-one-
out or 10-fold cross-validation is used for validation. If you don't
have access to data sets with larger number of samples, you might
want to try out the ensemble feature selection and prediction
methods to increase robustness. Advanced users might consider to
combine similar data sets together based on cross-study normalizaton
techniques to increase the number of samples. The simplest approach
is to try out different feature selection and prediction methods and
see, whether a certain combination provides consistently lower
standard deviations (but be aware of the multiple testing problem!).
However, depending on the size and quality of your data, even very
sophisticated algorithms might fail to overcome or to sufficiently
alleviate this problem.
Close
|
 How to cite us
How to cite us webmaster@arraymining.net
webmaster@arraymining.net